
Lesson 13 Confocal Microscopy
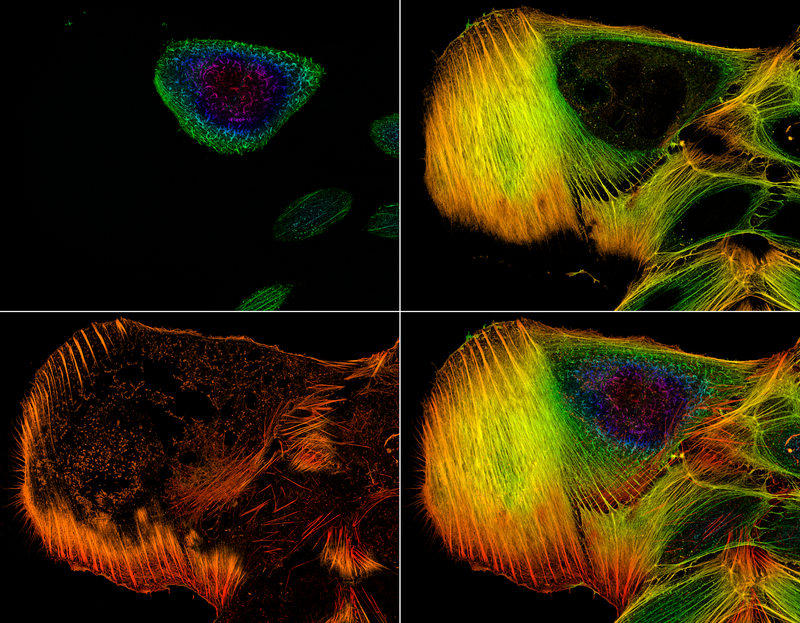
Confocal microscopy, most frequently confocal laser scanning microscopy (CLSM) or laser confocal scanning microscopy (LCSM), is an optical imaging technique for increasing optical resolution and contrast of a micrograph by means of using a spatial pinhole to block out-of-focus light in image formation.[1] Capturing multiple two-dimensional images at different depths in a sample enables the reconstruction of three-dimensional structures (a process known as optical sectioning) within an object. This technique is used extensively in the scientific and industrial communities and typical applications are in life sciences, semiconductor inspection and materials science.
Light travels through the sample under a conventional microscope as far into the specimen as it can penetrate, while a confocal microscope only focuses a smaller beam of light at one narrow depth level at a time. The CLSM achieves a controlled and highly limited depth of focus. The principle of confocal imaging was patented in 1957 by Marvin Minsky[2] and aims to overcome some limitations of traditional wide-field fluorescence microscopes.[3] In a conventional (i.e., wide-field) fluorescence microscope, the entire specimen is flooded evenly in light from a light source. All parts of the sample can be excited at the same time and the resulting fluorescence is detected by the microscope's photodetector or camera including a large unfocused background part. In contrast, a confocal microscope uses point illumination (see Point Spread Function) and a pinhole in an optically conjugate plane in front of the detector to eliminate out-of-focus signal – the name "confocal" stems from this configuration. As only light produced by fluorescence very close to the focal plane can be detected, the image's optical resolution, particularly in the sample depth direction, is much better than that of wide-field microscopes. However, as much of the light from sample fluorescence is blocked at the pinhole, this increased resolution is at the cost of decreased signal intensity – so long exposures are often required. To offset this drop in signal after the pinhole, the light intensity is detected by a sensitive detector, usually a photomultiplier tube (PMT) or avalanche photodiode, transforming the light signal into an electrical one.[4]
As only one point in the sample is illuminated at a time, 2D or 3D imaging requires scanning over a regular raster (i.e. a rectangular pattern of parallel scanning lines) in the specimen. The beam is scanned across the sample in the horizontal plane by using one or more (servo controlled) oscillating mirrors. This scanning method usually has a low reaction latency and the scan speed can be varied. Slower scans provide a better signal-to-noise ratio, resulting in better contrast.
The achievable thickness of the focal plane is defined mostly by the wavelength of the used light divided by the numerical aperture of the objective lens, but also by the optical properties of the specimen. The thin optical sectioning possible makes these types of microscopes particularly good at 3D imaging and surface profiling of samples.
Successive slices make up a 'z-stack', which can either be processed to create a 3D image, or it is merged into a 2D stack (predominately the maximum pixel intensity is taken, other common methods include using the standard deviation or summing the pixels).
Confocal microscopy provides the capacity for direct, noninvasive, serial optical sectioning of intact, thick, living specimens with a minimum of sample preparation as well as a marginal improvement in lateral resolution compared to wide-field microscopy.[4] Biological samples are often treated with fluorescent dyes to make selected objects visible. However, the actual dye concentration can be low to minimize the disturbance of biological systems: some instruments can track single fluorescent molecules. Also, transgenic techniques can create organisms that produce their own fluorescent chimeric molecules (such as a fusion of GFP, green fluorescent protein with the protein of interest). Confocal microscopes work on the principle of point excitation in the specimen (diffraction limited spot) and point detection of the resulting fluorescent signal. A pinhole at the detector provides a physical barrier that blocks out-of-focus fluorescence. Only the in-focus, or central spot of the Airy disk, is recorded. Raster scanning the specimen one point at a time permits thin optical sections to be collected by simply changing the z-focus. The resulting images can be stacked to produce a 3D image of the specimen. Four types of confocal microscopes are commercially available:
Confocal laser scanning microscopes use multiple mirrors (typically 2 or 3 scanning linearly along the x- and the y- axes) to scan the laser across the sample and "descan" the image across a fixed pinhole and detector.
Spinning-disk (Nipkow disk) confocal microscopes use a series of moving pinholes on a disc to scan spots of light. Since a series of pinholes scans an area in parallel, each pinhole is allowed to hover over a specific area for a longer amount of time thereby reducing the excitation energy needed to illuminate a sample when compared to laser scanning microscopes. Decreased excitation energy reduces phototoxicity and photobleaching of a sample often making it the preferred system for imaging live cells or organisms.
Microlens enhanced or dual spinning-disk confocal microscopes work under the same principles as spinning-disk confocal microscopes except a second spinning-disk containing micro-lenses is placed before the spinning-disk containing the pinholes. Every pinhole has an associated microlens. The micro-lenses act to capture a broad band of light and focus it into each pinhole significantly increasing the amount of light directed into each pinhole and reducing the amount of light blocked by the spinning-disk. Microlens enhanced confocal microscopes are therefore significantly more sensitive than standard spinning-disk systems. Yokogawa Electric invented this technology in 1992.[5]
Programmable array microscopes (PAM) use an electronically controlled spatial light modulator (SLM) that produces a set of moving pinholes. The SLM is a device containing an array of pixels with some property (opacity, reflectivity or optical rotation) of the individual pixels that can be adjusted electronically. The SLM contains microelectromechanical mirrors or liquid crystal components. The image is usually acquired by a charge coupled device (CCD) camera.
Each of these classes of confocal microscope have particular advantages and disadvantages. Most systems are either optimized for recording speed (i.e. video capture) or high spatial resolution. Confocal laser scanning microscopes can have a programmable sampling density and very high resolutions while Nipkow and PAM use a fixed sampling density defined by the camera's resolution. Imaging frame rates are typically slower for single point laser scanning systems than spinning-disk or PAM systems. Commercial spinning-disk confocal microscopes achieve frame rates of over 50 per second a desirable feature for dynamic observations such as live cell imaging.
In practice, Nipkow and PAM allow multiple pinholes scanning the same area in parallel as long as the pinholes are sufficiently far apart.
Cutting-edge development of confocal laser scanning microscopy now allows better than standard video rate (60 frames per second) imaging by using multiple microelectromechanical scanning mirrors.
Confocal X-ray fluorescence imaging is a newer technique that allows control over depth, in addition to horizontal and vertical aiming, for example, when analyzing buried layers in a painting.


Download Files
{kind=link}
- Lesson Plan, Books, Course outcome, Key dates etc
- Lesson 1 Introduction
- Lesson 2 History, Scope of Microbiology
- Lesson 3 History of Microbiology
- Lesson 4 Prokaryotes Structure
- Lesson 5 Prokaryotes Structure
- Lesson 6 Taxonomy
- Lesson 7 Taxonomy 2
- Lesson 8 Bacterial Growth
- Lesson 9 Bacterial Growth 2
- Lesson 10 Bacterial Growth 3
- Lesson 11 In-vitro cultivation of Bacteria
- Lesson 12 Microscopy
- Lesson 13 Confocal Microscopy
- Lesson 14 Staining Techniques in MIcrobiology
- Lesson 15 Electron Microscopy 1
- Lesson 16 Electron Microscopy 2
- Chapters 17
- Department Animal Sciences
- Teacher
Dr. Imtiaz Hussain
